


Presentation. A 38-year-old asymptomatic woman was referred to the retina service for fundus changes. Her history was significant for Lynch syndrome (autosomal dominant inheritable cancer syndrome, formerly referred to as hereditary nonpolyposis colorectal cancer). She had previously been diagnosed with adenocarcinoma of the colon and had undergone colon resection, chemotherapy, and radiation 2 years prior; she was in clinical remission. Her family history was remarkable for the same syndrome in her grandmother, mother, and brother, all of whom had died of metastatic colon cancer.
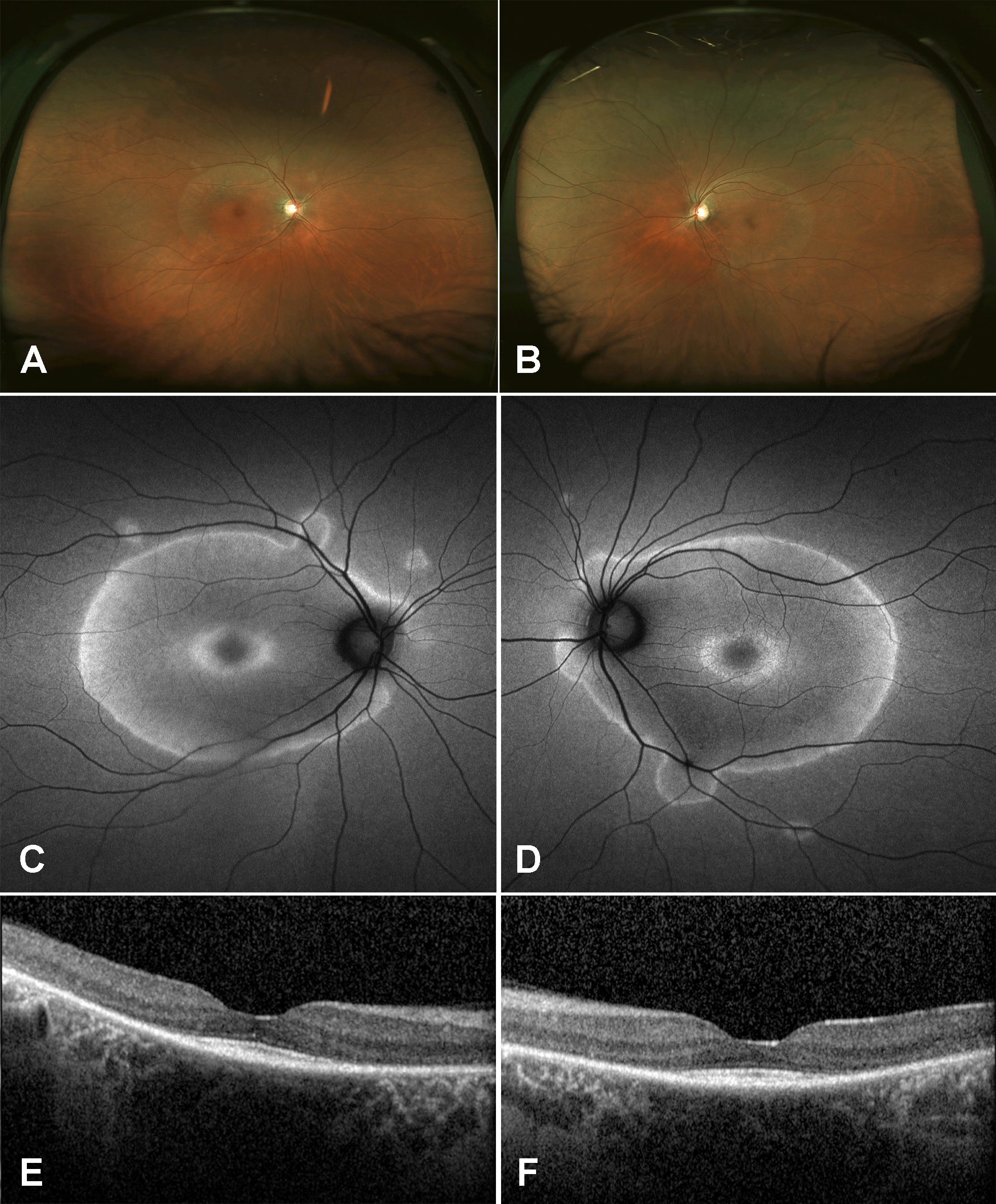
Figure 1 | Fundus photographs of the right eye (A) and left eye (B) showed perivascular RPE changes extending into the macula. Fundus autofluorescence of the right eye (C) and left eye (D) exhibited perivascular hyperautofluorescence with geographic borders. SD-OCT of the right eye (E) and left eye (F) revealed severe RPE and photoreceptor atrophy with foveal sparing.
Examination. The patient’s vision was 20/20 in both eyes with normal IOP, normal motility, and constricted confrontational visual fields. The anterior segment was normal. Dilated fundus examination revealed extensive perivascular retinal pigment epithelium (RPE) atrophy and mild vascular narrowing. There were no anterior chamber or vitreous cells. The patient’s fundus photos, fundus autofluorescence, and spectral-domain optical coherence tomography (SD-OCT) demonstrated changes consistent with extensive perivascular RPE and outer retinal atrophy (Figure 1). Thirty-two Humphrey visual fields (HVF) showed bilateral central scotomas (Figure 2). Full-field electroretinogram (ERG) demonstrated a severely diminished a-wave and b-wave under all scotopic conditions consistent with poor rod function (Figure 3). Under photopic conditions, there was also a severely diminished a-wave and b-wave consistent with poor cone function (Figure 3). The photopic 30-Hz flicker peak amplitude was 75 uV in the right eye and 95 uV in the left eye (normal: 108 ±39). The scotopic rod-specific b-wave amplitude was 167 uV in the right eye and 185 uV in the left eye (normal: 199 ±136).
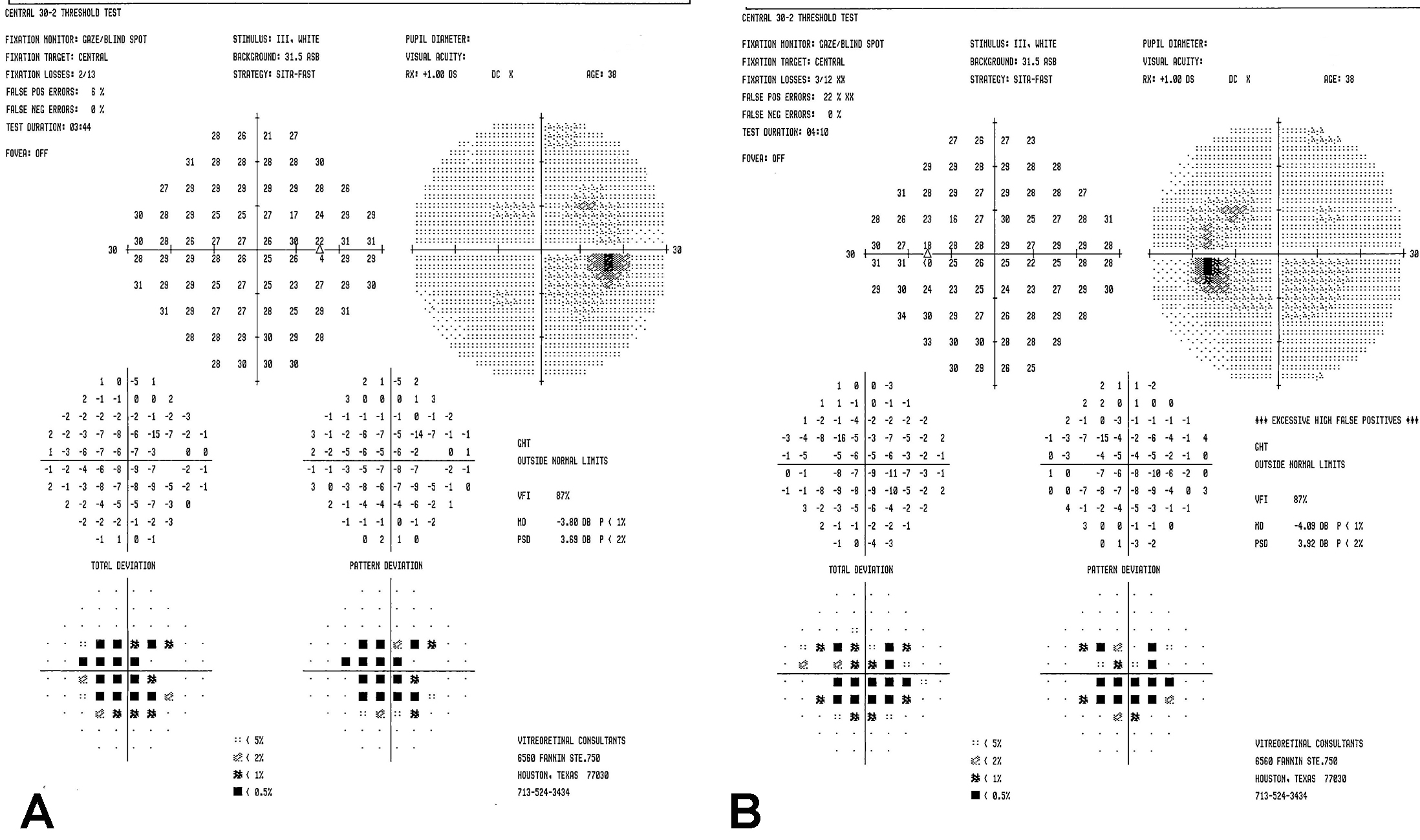
Figure 2 | Humphrey visual field in the right eye (A) and left eye (B) demonstrated central scotomas.
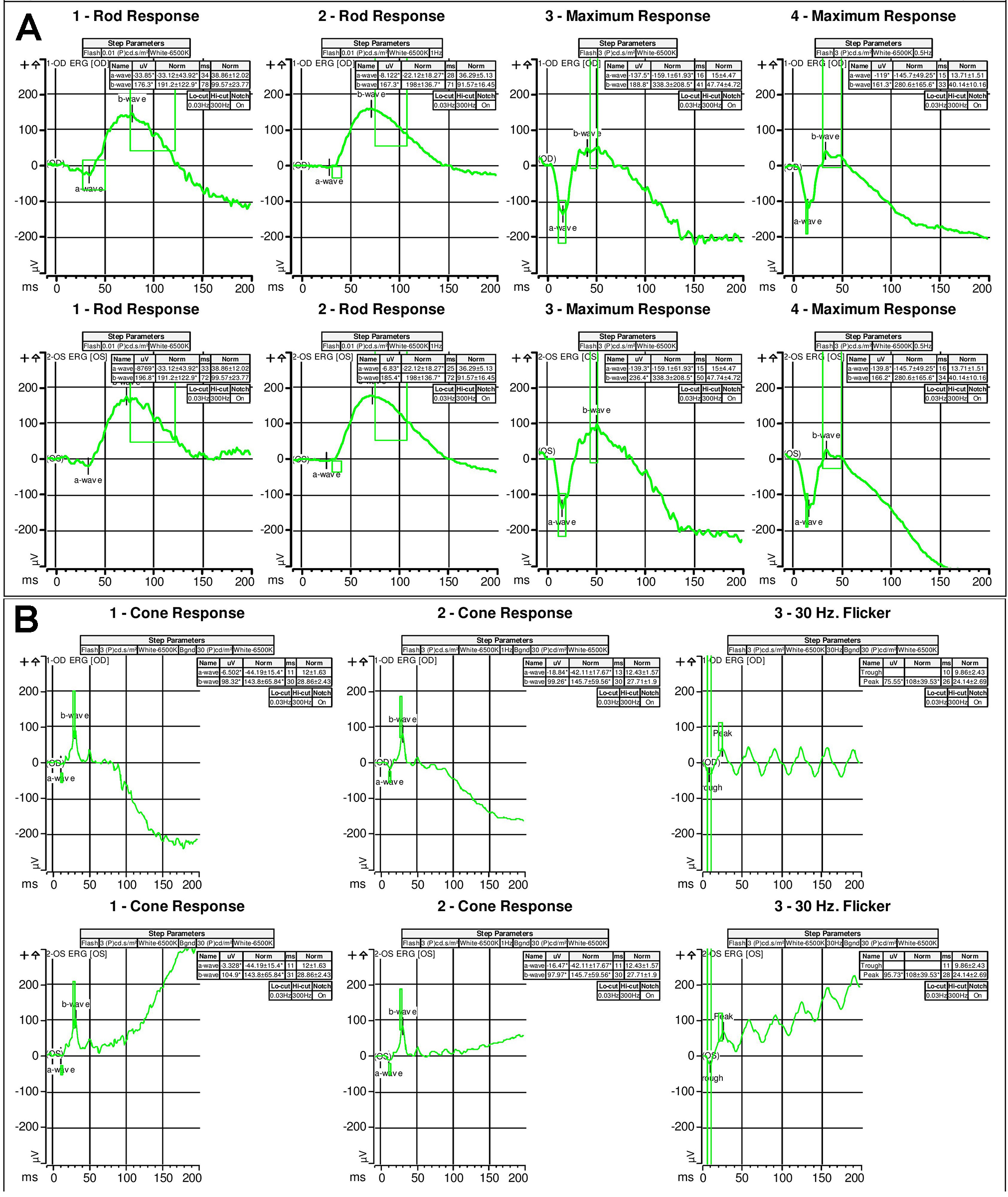
Figure 3 | Full-field scotopic electroretinogram (A) and photopic electroretinogram (B) illustrated severely diminished rod and cone function.
Diagnosis.To confirm the diagnosis, Western blot testing was ordered to assess for retinal autoantibodies and was positive for autoantibodies against a 22-kDa protein, 40-kDa protein (aldolase), and a 42-kDa protein. Although there were no autoantibodies against recoverin, the 23-kDa protein most commonly associated with cancer, the diagnosis of cancer-associated retinopathy (CAR) was made, given the patient’s clinical picture, history of cancer, and presence of retinal autoantibodies against multiple retinal proteins.
CAR is a rare paraneoplastic syndrome in which retinal autoantibodies directed at normal retinal proteins cause severe photoreceptor and RPE atrophy, resulting in progressive, relentless vision loss.1 Patients can develop the syndrome after receiving a cancer diagnosis and having been treated; during cancer treatment; or, in rare cases, years before a cancer is identified. The diagnosis is difficult to make, as early fundus examination typically reveals minimal fundus changes. Over time, vascular attenuation, RPE changes, and visual field changes become more apparent. The differential diagnosis can include autoimmune retinopathy, drug toxicity, retinitis pigmentosa and other degenerative retinal diseases, postinfectious changes, and other rare causes of RPE and photoreceptor loss.
Treatment. There is no standard therapy for the disease, and patients often have a poor overall prognosis due to the underlying cancer diagnosis.2 Published treatment approaches have included immunosuppression such as steroids (intravenous, oral, sub-Tenon, intravitreal) and immune-modulating therapies such as cyclosporine, azathioprine, mycophenolate, methotrexate, rituximab, and alemtuzumab. Other immune-modulating approaches have included intravenous immune globulin and anti-oxidant vitamins. None of these therapies has ever been studied in a large-scale prospective trial.
In collaboration with our hematology team, this patient was treated with five sessions, scheduled every 3 to 4 months, of plasma exchange, an immune-modulating treatment performed with a temporary dual-lumen venous catheter in which the patient’s plasma is removed and replaced with donor plasma or saline with albumin. This treatment approach had been previously published in a small series of patients with another paraneoplastic syndrome, bilateral diffuse uveal melanocytic proliferation,3 but never in CAR.
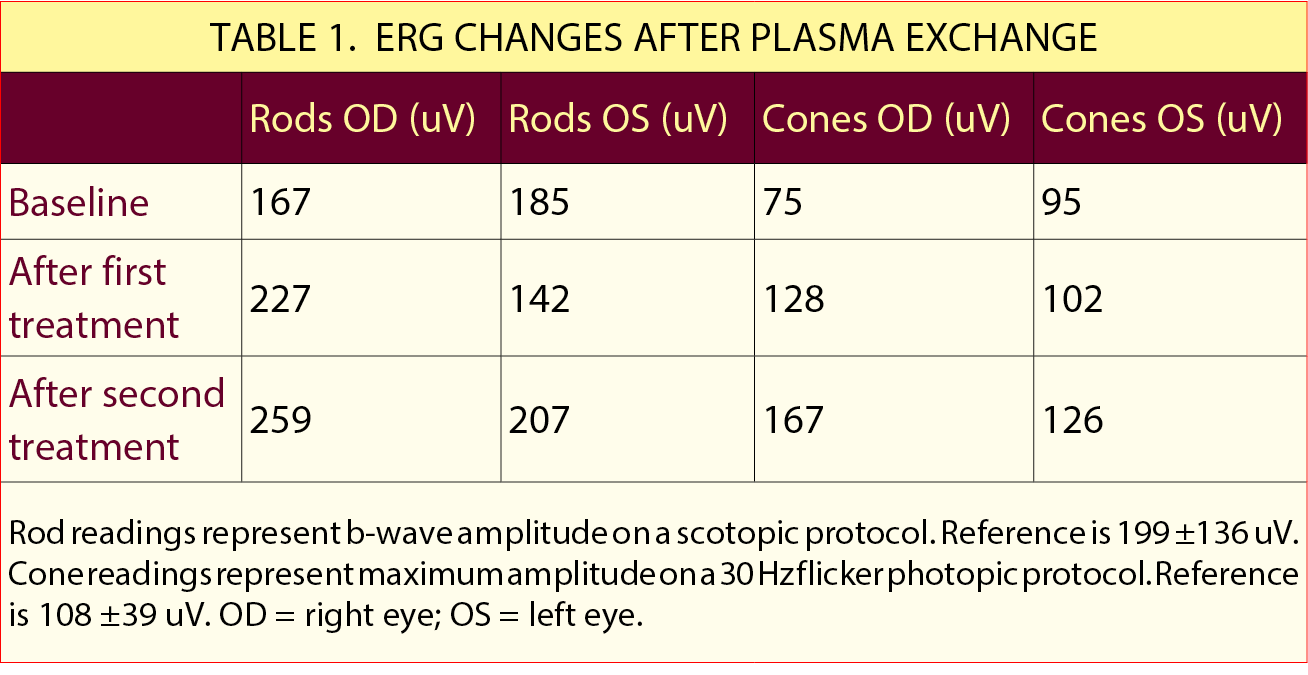
Outcome. The efficacy of the treatment was monitored based on the patient’s ERG response, which recovered after the first cycle and demonstrated a sustained response over two additional cycles. By the end of the third cycle, the scotopic b-wave amplitude was 259 uV in the right eye and 207 uV in the left eye (normal: 199 ±136). The scotopic rod-specific b-wave amplitude was 167 uV in the right eye and 126 uV in the left eye (normal: 108 ±39; Table 1). The patient reported a dramatic subjective improvement in vision, although the fundus photographic findings, SD-OCT, and HVF deficits remained unchanged.
On the Case is brought to you by The University of Texas Health Science Center at Houston (UTHealth) and Paxos Scope (DigiSight).
1. Goldstein SM, Syed NA, Milam AH, et al. Cancer-associated retinopathy. Arch Ophthalmol. 1999;117(12):1641-1645.
2. Chan JW. Paraneoplastic retinopathies and optic neuropathies. Surv Ophthalmol. 2003;48(1):12-38.
3. Jansen JC, Van Calster J, Pulido JS, et al. Early diagnosis and successful treatment of paraneoplastic melanocytic proliferation. Br J Ophthalmol. 2015;99(7):943-948.


